NNadir
NNadir's JournalH. Holden Thorpe: "This may be the most shameful moment in the history of U.S. science policy."
This week's editorial in the journal Science, written by the Editor in Chief of this prestigious scientific journal, comments on Trump's decision to lie to the American people about Covid-19, thus killing and severely injuring large numbers of them:
EDITORIAL: Trump lied about science (H. Holden Thorp Editor-in-Chief, Science Journals. Science 18 Sep 2020: Vol. 369, Issue 6510, pp. 1409)
Over the years, this page has commented on the scientific foibles of U.S. presidents. Inadequate action on climate change and environmental degradation during both Republican and Democratic administrations have been criticized frequently. Editorials have bemoaned endorsements by presidents on teaching intelligent design, creationism, and other antiscience in public schools. These matters are still important. But now, a U.S. president has deliberately lied about science in a way that was imminently dangerous to human health and directly led to widespread deaths of Americans.
This may be the most shameful moment in the history of U.S. science policy...
...Trump also knew that the virus could be deadly for young people. “It's not just old, older,” he told Woodward on 19 March. “Young people, too, plenty of young people.” Yet, he has insisted that schools and universities reopen and that college football should resume. He recently added to his advisory team Scott Atlas—a neuroradiologist with no expertise in epidemiology...
...Monuments in Washington, D.C., have chiseled into them words spoken by real leaders during crises. “Confidence,” said Franklin Roosevelt, “thrives on honesty, on honor, on the sacredness of obligations, on faithful protection and on unselfish performance.”
We can be thankful that science has embraced these words. Researchers are tirelessly developing vaccines and investigating the origins of the virus so that future pandemics may be prevented. Health care workers have braved exposure to treat COVID-19 patients and reduce the death rate; many of these frontline workers have become infected, and some have died in these acts of courage. These individuals embody Roosevelt's call to faithful protection and unselfish performance.
They have seen neither quality exhibited by their president and his coconspirators. Trump was not clueless, and he was not ignoring the briefings. Listen to his own words. Trump lied, plain and simple.
Scientists act as if they are above and removed from politics at their own risk, and as pointed out here, at a risk to science itself.
My fellow married old farts: Do you love to look at pictures of your spouse when he/she was young?
My wife was 22 when I married her, just turning 21 when we moved in together.
I love her very much decades later, in ways that matter far more than when it a big part of my love for her was involved with the fact that she was incredibly sexy, but I still find myself looking at her when she was young, when that odd thing was so important.
I don't know what I'm trying to see in those pictures. What am I trying to see, what is the yearning? In a way, it almost feels perverse.
Of course, no real disrespect, but now 21 year old people seem so much like kids - smart kids to be sure, kids perhaps wiser than I was at 21, better kids than I was at 21 to be sure, fine kids - but they're still just kids.
It is strange, though, because it occurs to me that as I have always seen my wife as a full woman, when I was just living with her, when we first married, but was she just a kid? Am I really looking at a full grown woman when I look at those pictures? Was there something of "just a girl" there?
There are many things that are wonderful about growing old, powerful perceptions, but many things that seem more mysterious than ever and in some sense disturbing: The challenge of the veracity of memory is one of them.
I took a day off from work, and spent it with her, walking around downtown Princeton, masks off for lunch on a sidewalk, and suddenly I'm struck by a kind of wonder...how I wonder...how did the time go away?
Noninvasive Assessment of Traumatic Brain Injury by LC/MS/MS Determination of 8 Urinary Metabolites.
The paper I'll discuss in this post is this one: Simultaneous Determination of Eight Urinary Metabolites by HPLC-MS/MS for Noninvasive Assessment of Traumatic Brain Injury (Shi et al., J. Am. Soc. Mass Spectrom. 2020, 31, 9, 1910–1917).
When your Doctor orders blood or urine tests to obtain information about your state of health, the general term for the analyte being measured is a "biomarker." Blood levels of glucose, or A1C, for example are biomarkers for diabetes, cholesterol, a biomarker for heart health. Quietly, without much fanfare, there has been a revolution going on in the determination of biomarkers with increasing sensitivity. To some extent, this revolution - at least originally - was involved in something known as "competitive binding assays" or "ligand binding assays." When I was a kid I used to make reagents for these assays that were labeled with radioactivity, "RIA" reagents, "radioimmunoassay" reagents; almost no one uses this technology anymore, it has been supplanted by ELISA, enzyme-linked immunosorbent assays, in which the radiation detected is of much lower energy than is provided by radioactive materials: It is the radiation we know as "visible light." I expect that within a decade, much of the ELISA testing will go the way of RIA, supplanted by mass spectrometry, or at least in tandem with mass spectrometry.
Mass spectrometry, in general, falls into two classes, "triple quads" which provide high sensitivity for the "work a day" world - including commercial medical laboratories, and in research, high resolution (where resolution refers tp mass accuracy) mass spectrometry, which can be of several types, "orbitrap," "time of flight" as well as the much rarer but incredibly accurate "Fourier Transform Ion Cyclotron Resonance Mass Spectrometry (FT-ICRMS).
A recent trend has been to approach combining high resolution mass specs with high sensitivity; this trend is only going to continue.
In the absence of validated biomarker analysis, diagnosis can involve considerable risk. Personally, I have an unusual EKG. Since I was a young man, doctors have often interpreted my EKG as implying that I have had - or am having - a heart attack. I've actually been hospitalized several times because of this, and once was briefly admitted to ICU, despite the fact that I have not had a heart attack. The second time I went through this process, my physician recommended that I have an angiogram, which involves inserting a catheter directly into the heart. While the test is very cool - you're on a local anesthetic and can actually watch the chambers of your beating heart on a television screen along with the doctor - it also is fairly risky. I had to sign all sorts of releases stating that I was aware that the test could either severely injury me or kill me. (There is a non-invasive biomarker test, "cardiac enzymes," troponins, that can indicate an active heart attack - but the damage done by previous heart attacks - I've had this test a number of times.
The situation with respect to the brain is even more complicated. Although in recent years many technologies have been developed for brain imagining, since the brain is an organ where many problems, for example Alzheimer's, actually take place on the cellular or molecular level.
The instrument utilized in this paper is certainly not a "state of the art" instrument, but it is a solid instrument that was widely in use ten or fifteen years ago, and was very popular, bordering on something of a scientific "cult" - the Sciex API 4000, a triple quad instrument. This lab, at the University of Missouri is probably not funded enough to drop half a million dollars regularly on the latest and greatest mass spectrometer every two or three years. Nevertheless, it's good work, which can lead people - some of whom will have more advanced instrumentation - to have something on which to build.
From the introduction:
Molecular biomarkers can provide additional biological information about concussive injuries that can complement conventional neurological assessments and medical imaging. Prior efforts to identify TBI biomarkers have noticed novel associations between several proteins found in cerebrospinal fluid (CSF) and serum to clinical outcomes in patients with TBI.(9) For example, glial S100?, which is involved in low affinity calcium binding in astrocytes, has been connected to astrocyte stress and death.(10) Glial fibrillary acidic protein (GFAP) has also been extensively studied owing to its function as an intermediate filament protein in astrocytes, where increased serum levels have been associated with astrocyte damage.(11,12) Additionally, serum levels of neuronal, specifically axonal, damage indicators, including neuron-specific enolase,(13) cleaved tau protein,(14) and ubiquitin C-terminal hydrolase,(15,16) have been correlated with poor clinical outcomes.
It is possible to obtain "CSF" for medical tests - and it is also possible to detect proteins by mass spec, either intact, with high resolution MS or by digestion with triple quads - but obtaining CSF is a somewhat involved and possibly traumatic enterprise. Urine is easier to get, obviously.
The authors rationalize their choice of biomarkers - all of which are small largely endogenous molecules. (Endogenous molecules are somewhat more difficult to analyze than molecules - such as drugs - that do not occur naturally in the body simply because it is difficult to procure blanks lacking these molecules but also containing possible interfering species.
....(17,22?24) Furthermore, excess oxidative stress occurs as a result of mitochondrial damage and altered oxidative metabolism, which has prompted interest to explore oxidative stress metabolites, most notably N-acetylaspartic acid (NAA)(25,26) and F2?-isoprostane,(27,28) as potential TBI biomarkers. This complex pathophysiological cascade invokes distinct metabolic processes that may therefore be probed to assess the extent and nature of the injury.(17,18,29) We have therefore proposed a novel combination of these interesting metabolites, shown in Figure 1, alongside several established protein biomarkers, to study potential patterns and differences in TBI samples. This panel, composed of a combination of excitatory neurotransmitters, glycolytic intermediates, and oxidative stress indicators, represents broad and diversified potential biomarkers of neurological processes involved in TBI...
...While cerebrospinal fluid has conventionally served as the gold standard for brain metabolite quantitation, owing to its direct contact with the extracellular matrix of the brain,(30) urine and serum present minimally invasive specimen types that can be readily obtained in the field. In particular, urine offers several advantages including large sample volumes, fewer sample pretreatment requirements, and noninvasive sample collection requiring no medical expertise. However, and to the best of our knowledge, a comprehensive panel of TBI biomarkers has yet to be developed in urine... ...In this study, we have therefore proposed a new technique for simultaneous determination of eight chemically diverse metabolites in urine without intensive sample preparation using high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS). This novel approach may provide timely supplemental characterization of traumatic brain injuries in the field...
Here are the biomarkers and their structures as reported in the paper, in one case incorrectly:

The caption:
The structure drawn for 5-hydroxindole acetic acid is missing the "5-hydroxy" and thus may generate a correction note in a subsequent issue of the journal. Probably the structures were drawn by a student.
What is noticable is that many of these molecules are generally ubiquitous in cells: Pyruvate is a constituent of the citric acid (Kreb's) cycle which takes place in mitochondria. Glutamic acid is an amino acid found in practically every protein - the monosodium salt is the famous "MSG" which upsets certain people eating Chinese food. Methionine sulfoxide is a derivative of methionine, a common amino acid in many proteins, wherein the methionine is oxidized to its sulfoxide as part of normal physiological processes, for example methylation reactions.
The simplicity of these molecules makes their analysis significantly more challenging than it is for complex molecules. This said, the F2?-isoprostane is challenging because it belongs to a class of lipid molecules known as prostaglandins, themselves a subset of molecules called "eicosanoids," the metabolic products of highly unsaturated acids. (These molecules are important to inflammatory pathways involved in healing and in immune responses.) A problem with these classes of molecules is that different molecules, with different roles can have the same molecular weight - we call these "isobaric" molecules - with the result that since a mass spectrum depends on mass signals, and thus can give ambiguous and indeed, wrong results. (This possibility is not discussed in the paper, but presumably it was addressed in the laboratory.)
Here is the chromatogram of the analysis, which is actually in two dimensions, one being a mass related dimension ("mass transitions" in fragmentation, and the other in time, specifically retention time, the time it takes for the molecule to emerge from the separating chromatography column. The first is using reference standards:

The caption:
Figure 2. Representative overlaid XIC chromatogram of the eight metabolite standards prepared at 500 ?g/L in synthetic urine.
The second is a representative chromatogram:

The caption:
Here are tables of the method performance:

Here the authors report the LOD, limit of detection, rather than the more useful LOQ, limit of quantitation, but the reported signal to noise ratio is decent.
I have seen prostaglandin analysis that have LOQ's that are two orders of magnitude lower than what is reported here as an LOD, but this was very challenging to accomplish and involved far more advanced instrumentation than a Sciex API 4000.

Strictly put, the spike recovery for the prostaglandin, its accuracy, is out of range for a regulatory method, which may or may not be involved in interference, but it's not bad overall.
My snottiness aside, the method is pretty decent within its limits - it's an academic, not a regulatory method - and has a reasonably short run time for a method measuring 8 analytes simultaneously.
The conclusion is a little bit overstated, but not so much as to be entirely unreasonable:
It is notable that urine - despite its apparent simplicity - can be a difficult matrix for mass spec for a number of reasons, and the authors have done a very workman like job within the limits of their equipment base. It's nice work.
I trust you will have a safe, healthy, and enjoyable weekend.
I spent 3 1/2 hours last night listening to Amon Goeth's (Schindler's List) former slave talk...
...about her life as his slave.
(I have recently renewed study of this period because of am better understanding, because of our Nazi "President" how it happened.)
It was an interview as part of USC's Shoah project, which features living history videos. The video is the testimony of Helena Jonas Rosenzweig, one of "Schlinder's Jews," who actually lived in Goeth's house.
She was an articulate speaker with such overwhelming depth of humanity; she died in 2018, and interviewed when she was 71.
In the movie, Schinder's List, she was represented by a composite character, as she was one of two housekeeping slaves held by the vicious camp commandment, the war criminal Amon Goethe, who was executed by the Poles in 1946.
Zero to the start: The interview begins a few seconds after the test pattern.
Revealing Mechanistic Processes in Gas-Diffusion Electrodes During CO2 Reduction to CO/Formate.
The paper I'll discuss in this post is this one: Revealing Mechanistic Processes in Gas-Diffusion Electrodes During CO2 Reduction via Impedance Spectroscopy (Bienen et al., ACS Sustainable Chem. Eng. 2020, 8, 36, 13759–13768)
Electricity is decidedly not a primary source of energy, and as such, it is always a thermodynamically degraded form of energy. It is therefore intrinsically wasteful. Storing electricity - generally as chemical energy although there are limited opportunities to store it as gravitational energy or as compressed gases - further thermodynamically degrades electrical energy and also is intrinsically wasteful both at the point of storage and at the point of release. Batteries, for example, give off heat, charging and discharging. Despite vast enthusiasm for energy storage by the general public, and tens upon hundreds of thousands of papers written on this subject, all of this is generally true, as I often state in my posts in this space.
There is one caveat however that is possibly of some possible import - if one captures energy that would have been wasted anyway and stores it in a form that is usable - the thermodynamic losses, although there still will be losses, will be minimized. At the most extreme, the only form of primary energy is nuclear. The sun is famously a nuclear device, which drives the wind, provides light - which over hundreds of millions of years was converted into chemical energy, dangerous fossil fuels which we can't burn fast enough - and also into current inventories of biomass, including but not limited to food. Some of the very best minds of the 20th century learned how to harness nuclear energy in the form of fission, although strictly put, uranium and thorium and the elements made from them are actually stored energy from ancient supernovae. Indeed, geothermal energy, which is provided by heat released by nuclear decay is also stored energy from ancient supernovae, being released in a generally regular way, earthquakes and volcanoes not withstanding. Only tidal energy is seemingly divorced from the nuclear primacy.
Nevertheless in practical engineering terms, we tend to think of dangerous fossil fuels and related biomass as primary energy, light from the sun as primary energy, wind as primary energy, geothermal as primary energy, gravitational energy, which is used in tidal and hydroelectric systems (the latter still driven by the sun) as primary energy, and of course, nuclear fission as primary energy.
The paper listed at the outset of this post is about wasting energy to make electricity and then wasting even more of it to make chemical energy, in this case formate and carbon monoxide, the latter, via the "water gas" reaction, an equivalent of hydrogen.
Despite my contempt for the thermodynamics of electricity, I do sometimes imagine cases where it might be acceptable to waste some energy to make electricity, and even chemical energy via electricity.
To illustrate, I offer a diversion. Over the last week or so, at various times of the day, on various days of the week, I've been downloading the data and graphics on the CAISO systems status pages, in three areas, demand, supply, and emissions. CAISO monitors and oversees electricity in California, CAISO = CAlifornia Independent System Operators. They have these wonderful real time page with graphics and numbers that will tell you the state of affairs with respect to electricity at any moment of the day. It's here: CAISO Pages
Here are some graphics I downloaded a little while ago as of this writing, near the 17th hour of the day, Pacific Time:
Here is the demand for electricity in California; the shaded area is historical data for the day, the line beyond is the predicted electricity demand.

The 17th hour, 5 pm PST or PDT +/- an hour or two, as the case might be is consistently close to the peak demand for energy in California, weekends, work days, holidays, every day.
Most people know that the sun is "going down" around that time, and as a result the output of solar energy is declining. California is unique among those places invested in the "renewables will save us" fantasy, in that the output of solar energy can dominate, at times, the output of so called "renewable energy", at least around noon.
This graphic and data breaks down what percentage was around the 17th hour of September 15, 2020:

This graphic and data shows the data for the September 15, 2020 up to 17:10 (5:10 pm):

The absolutely flat grey line near the bottom, 2,241 MW, is California's last remaining nuclear reactor, Diablo Canyon, two nuclear reactors contained in a single building. From midnight until 7 am this morning, it was producing just about as much energy as all of the wind turbines, solar cells, biomass combustion plants (which almost certainly include garbage incinerators), and geothermal plants in the entire state.
The disturbing line, of course, is the "imports" line and the "natural gas" line.
Here are California's emissions, as of 17:10 (5:10 pm PDT).

Don't hold me to this, but my general impression is that if you were to average all the "per hour" emissions data I've seen over the course of pulling up and downloading 20 or 30 of these files, the average would be somewhere between 7,000 metric tons and 8,000 metric tons of carbon dioxide ruthlessly dumped by the State of California to provide electricity to its citizens.
There are 8,767.76 hours in a sideral year, suggesting that California dumps about 65,750,000 metric tons of carbon dioxide into the planetary atmosphere each year, this while being "green" and regularly passing, going back to the wonderful time I lived in that State, now decades ago, "by 2000" and "by 2020" and "by 2050" and "by 2060" energy bills claiming that the state will be "green," at least as they imagine "green."
To be honest, my contempt aside, this actually isn't bad, and on their emissions page they have one of those innumerate "percent talk" statements about how great they're doing on emissions.
The reality is however, that all this talk, half a century of it, has not addressed climate change. California, and Oregon, and Washington, recently Australia, this week the Pantanal, are all burning, and powerful hurricanes slice through the Southeastern and Northeastern United States like coupled freight trains.
We needed to be releasing zero carbon dioxide into the atmosphere to generate electricity, and we needed to be doing it years ago.
I just went on the CAISO site again: Today's peak electricity demand took place at 17:18 this afternoon, at 35,971 MWe.
Diablo Canyon is not very impressive in terms of its thermodynamic efficiency with respect to its "primary" energy. Like most nuclear power plants designed in its era, it's around 33% efficient, atrocious really, but similar to all historic Rankine cycle plants. Modern gas powered plants, which are combined Brayton/Rankine cycle plants are closer, and sometimes exceed 50% efficiency.
The reality is, that today, to have provided all the electricity for today's peak demand, 35,971/2,241 would require 17 buildings the size of Diablo Canyon of Diablo Canyon's precise design - which is, by the way, going to be shut in three or four years, because it's "too dangerous" in the minds of people who can't think very well, but the huge tracts of the state burning each year is not "too dangerous."
Of course, at around 3:40 am on September 15, 2020, the demand for electricity in California was less than 22,000 MWe, and the bulk of the putative imaginary nuclear power plants would not be necessary.
In general, my thermodynamic sense is that it is stupid to generate electricity to create potential chemical energy. It is much smarter to use direct thermal to chemical energy conversion if one were to store energy.
However, all chemical processes require cooling, and the rules of process intensification, which are rules to derive the most usable energy per unit of primary energy generated, can conveniently convert thermal energy to electricity with Rankin or Brayton or Stirling heat engines, and storing this mechanical energy may be justifiably be done with an electricity intermediate.
The chemical process in the paper that I promised to discuss at the outset uses carbon dioxide as a feedstock; if this carbon dioxide is obtained from the waste dump into which we've transformed our planetary atmosphere, that can help clean it up.
Here is the cartoon associated with the paper's abstract, which can be read at the link:

From the introduction to the paper:
This work focuses on the tin-catalyzed conversion of CO2 to obtain formate as the target product. Typically, CO2 is not solely converted to formate (eq 1) on tin catalysts but also to carbon monoxide to a lower extent (eq 2), while the aqueous electrolyte is reduced to hydrogen (eq 3) in a parasitic side reaction, reducing the charge efficiency with respect to formate formation, according to the following equations (1) (2) (3)

Formate can be used as a deicing agent or drilling fluid as well as for tanning and silage when protonated to formic acid.(9) In a proof-of-concept study, it was also shown that formate obtained via CO2 reduction reaction (CO2RR) can be utilized as energy carrier and fed into a direct formate fuel cell to produce electricity or after catalytic decomposition to hydrogen with subsequent re-electrification in a typical polymer electrolyte membrane fuel cell.(10,11)
Unfortunately, the conversion of dissolved CO2 on planar electrodes is limited to a maximum current density of well below 10 mA cm–2 because of mass transport constraints evoked by the low solubility of CO2 in the aqueous electrolyte (33 mM L–1 in H2O, 25 °C, 1 atm) and the diffusion of dissolved CO2 from the bulk electrolyte to the electrode surface.(7,12,13) This limitation can be circumvented by the use of so-called gas-diffusion electrodes (GDEs) providing a porous architecture and intensifying the contact between the gas-, liquid-, and solid phases. Their use entails a substantial increase in the number of active sites while the diffusion length of dissolved CO2 to the catalyst surface is reduced. Accordingly, gaseous CO2 can be employed as the substrate and, due to the above effects; the macroscopic mass transport of the reactant is substantially accelerated.(14?16) As a result, the achievable current densities when using GDEs for CO2 electrolysis can be increased by more than an order of magnitude compared to planar electrodes without sacrificing selectivity toward CO2RR products.(17?19) The so far achieved current densities which are already on industrially relevant orders are the reason why GDEs have gained increasing interest in recent years for the investigation of CO2 electrolysis systems.(15,19?21) Nevertheless, long-term stability of these GDEs is an under-represented research topic in the literature. Besides potential catalyst degradation, GDEs might suffer from a change of their hydrophobic properties over time, resulting in flooding, efficiency losses, and in a shift of the product selectivity toward the undesired hydrogen evolution.(22,23) In that respect, electrochemical impedance spectroscopy (EIS) is a powerful tool to deconvolute contributions of specific physical and (electro)chemical processes to the overall resistance during operation of electrochemical devices. The knowledge which process (e.g., charge-transfer, mass transport of reactants) determines the polarization resistance gives valuable insights required for the rational optimization of the employed GDEs and can aid in revealing possible degradation mechanisms. To use EIS as an electrochemical diagnostic tool, it is crucial to know which physical phenomena are observed in the measured impedance spectra...
About their approach using Electrochemical Impedence Spectroscopy:
Before discussing the results of the EIS measurement, it is useful to get a general understanding of CO2 reduction employing the GDEs manufactured in our laboratory. A polarization curve for the GDE operated with 100 vol % CO2 in 1.0 M KOH at 30 °C is depicted in Figure 1a. The curve was recorded via a stepwise increase of the current and logging the corresponding potential after 15 min at each value. At the maximum current, ?400 mA cm–2, the potential is approx. ?725 mV with a FE for CO and H2 of about 12 and 5%, respectively. The remaining 83% is attributed to the production of formate. A slight scattering of the potentials at higher currents is induced due to the gas evolution at the GDE surface making a precise potential determination impossible. The observed FEs for CO (?12%) and H2 (?5%) remain nearly constant for potentials of ?450 mV to ?725 mV. This fact indicates that even at a current density of ?400 mA cm–2, no mass transport limitation for CO2 is observed because otherwise the FE for H2 would increase. For 1 and 0.5 mA cm–2, the FEs were not determined because the quantification error would be too high due to the low amount of charge involved.
Figure 1:

The caption:
FE is Faradaic Efficiency, which is a function of how many electrons participate in the desired reaction as opposed to side reactions. It is not equivalent to thermodynamic efficiency.
More commentary:
In Figure 2, the current- and temperature-dependent Nyquist and imaginary impedance versus frequency plots are shown. The obtained FEs for H2 and CO during the EIS measurements are in good agreement with the observed values for the FE acquired during the measurement of the polarization curve. The medium-frequency process around 20 Hz shows a distinct dependence on the applied current. A higher current leads to an exponential decrease of the corresponding medium-frequency resistance (cf. Figure 2a). Additionally, Figure 2b reveals that the characteristic frequency of this resistance shifts to higher values when increasing the current. The exact same trends can be observed for the medium-frequency process for an increase of temperature (cf. Figure 2c,d). This temperature- and current-dependent behavior is commonly associated with a charge-transfer process and strongly indicates that the medium-frequency process displays a charge-transfer reaction.(34,37) A process in the low-frequency region (on the right in the Nyquist plot) can be observed for the lowest current density of ?25 mA cm–2 (cf. Figure 2a).
Figure 2:

Some additional graphics:

The caption:

The caption:
Figure 4. (a) Impedance spectra recorded during electrolysis for varying CO2 volume fractions in the feed gas and (b) time-dependent behavior of the impedance spectra for the operation with 20 and 10 vol % CO2 in the feed gas.
Some more commentary:
In the literature, there is still ongoing debate about whether CO2 (aq) or HCO3– (aq) is the active species during the rate-limiting step of the CO2RR.(8,41?44) To gain further insights into this matter, we will present experiments related to kinetic isotope effects (KIEs) in the following in which D2O is employed as solvent for the electrolyte. Processes which are affected by hydrogen atoms are expected to be slowed down when substituting hydrogen with heavier deuterium which should shed light on the species involved in the reaction.(45)
The changes when substituting H2O with D2O become evident comparing the data in the imaginary part versus frequency part representation of the results. These plots for pure N2 and CO2 gas feed in H2O- and D2O-based KOH electrolytes are depicted in Figure 5 and partially reveal significant differences in the characteristic frequencies of the processes. The measured spectrum for the operation of the GDE with pure N2 will be determined by HER only and should be influenced when using D2O as solvent and is used as benchmark comparison to demonstrate how a spectrum changes when it is affected by the substitution of hydrogen with deuterium. Indeed, the characteristic frequency, or in other words the velocity of the observed charge-transfer process, is reduced from 6.6 to 1.5 Hz when using D2O as solvent (cf. Figure 5a). The peak height which is in first proximity proportional to the resistance is increased, and the iR corrected potential also increases from ?1003 to ?1072 mV. Unsurprisingly, the ratio of the deuterium and hydrogen gas volume fraction (0.73:1.0) in the product gas stream agreed very well with the ratio of the corresponding thermal conductivities (0.75:1.0, ?D2 = 138 mW K–1 cm–1, ?D2 = 185 mW K–1 cm–1)(46)...

The caption:
Figure 5. Imaginary part vs frequency plots obtained from EIS measurements during electrolysis using pure (a) N2 and (b) CO2 as feed gas and H2O- or D2O-based 1.0 M KOH as the electrolyte.
An excerpt from the conclusion:
I personally love radicals, and that carbon dioxide radical is a very interesting species.
I'm not particularly great with electrochemistry, but I rather enjoyed the effort to get my head around this paper.
It also gave me a chance to ruminate on when the deliberate generation, for storage, of the thermodynamically degraded (but useful) form of energy, electricity, is acceptable for energy storage.
I trust you will have a safe and pleasant day tomorrow and will sleep well tonight.
I wish I had a river so long...
Power ideals and beauty fading in everyone's hand...
Hidden Perils of Lead in the Lab: Containing...Decontaminating Lead in Perovskite Research.
The paper I'll discuss in this post is this one: Hidden Perils of Lead in the Lab: Guidelines for Containing, Monitoring, and Decontaminating Lead in the Context of Perovskite Research (Michael Salvador,* Christopher E. Motter, and Iain McCulloch, Chem. Mater. 2020, 32, 17, 7141–7149)
When I was a kid, I worked a few years with radioactive iodine-125 to make reagents for an early and once popular form of competitive ligand binding assays; radioimmunoassay kits. The radioactive iodine came in the form of an iodide salt, and the procedure was to oxidize the iodine with a reagent called "Chloramine T," N-chlorotoluene sulfonamide, an oxidant that generates the electrophilic reagent chloroiodine and, inevitably, some free iodine, which can and does volatilize. The chloroiodine would react with aromatic rings - most typically on tyrosine in proteins, and the protein (or other standard) would be thus labeled and detectable at very low levels using a scintillating detector, but some would always escape from the test tubes in which the reactions were performed.
Although iodinations were conducted in the hood it was inevitable, especially because chromatography was generally conducted on benchtops in this lab, that surfaces in the lab would become contaminated with radioiodine, including floors and benchtops, and ultimately the result that scientists working in the lab would themselves become contaminated. Most everyone had a thyroid count after a few months, but one could minimize it by taking iodine supplements, something I encouraged among my peers.
The fun thing was that one could almost always see where and how much one was contaminated with the simple use of Geiger counter, and one could discover quite readily how effectively one's safety practices, gloving, lab coats, lead aprons were working. I had a lot of fun with this. Because I understood radiation better than most people in the lab - these were tools for biological assays after all - I generally took responsibility for handing clean up and waste disposal and I used this experience to learn a lot about contamination and decontamination, which proved useful throughout my career. (If one iodinates proteins in one's skin, one cannot wash it off of course, which is why gloves were always worn, but even these were never 100% effective.)
Of course, working with radioiodine - which is immediately detectable - is good practice for working with nasty metals, like, as is discussed in this paper, lead, which is not immediately detectable and thus is in many ways worse. The same is true of say, Covid-19 viral particles. I would recommend that anyone who is wearing gloves in situations of possible Covid exposure including exposure in the general public and who has not be trained in contamination and decontamination, take a good look at the figure I will post below.
It should be obvious that we have failed miserably to prevent the contamination of the entire planetary atmosphere with carbon dioxide (and for that matter aerosols of the neurotoxins lead and mercury released in coal burning) but that hasn't prevented humanity from doing the same thing over and over again and expecting a different result. Here I am referring to the quixotic effort to displace fossil fuels - although the focus has often drifted into a stupid and frankly dangerous effort to displace infinitely cleaner nuclear energy rather than fossil fuels - with so called "renewable energy," the most popular form being "solar energy."
The failed effort to address climate change with solar energy hasn't lacked for ever more elaborate schemes, and frankly, although I deplore the solar energy industry in general because of its most dangerous feature - it doesn't work to improve the environment - some interesting science has nonetheless resulted from the huge expense, some of which, unlike the solar industry itself, may someday have practical import.
The latest fad in solar research is in the area of perovskites, solids having the following molecular structure:
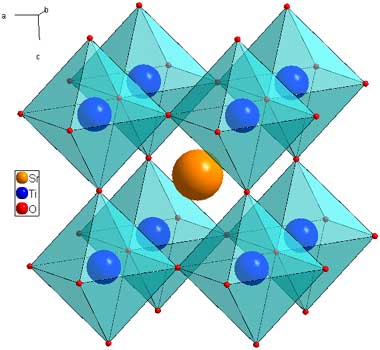
(This cartoon is found on the web pages of the Cava Lab at Princeton University.) One can see that these are octahedra embedded in cubic structures.
The most famous of the perovskites in the endless and continuously failed quest to make solar electricity produce energy on a scale that is meaningful are ternary compounds of cesium, iodine and lead. (Sometimes the cesium can be substituted with small organic ions.) There are actually people who believe that distributing lead for "distributed energy" would be a good idea.
I'm not kidding. The rationale is that "it's not as bad as coal burning" or "it's not as bad as car batteries" which is like saying "breast cancer is not as bad as pancreatic cancer."
Go figure.
Laboratory research is often dangerous and I know from experience that people can get quite cavalier about it. It's a bad idea to get cavalier. People's health can be impacted by the reagents with which they work.
Hence this paper is quite a useful reminder that, um, playing with lead base perovskites can be bad for you.
...Lead(II) salts are precursors of lead-based perovskite semiconductors. In a typical perovskite research lab, spillage of trace amounts of lead powder is unavoidable. Unlike a simple spill of a benign chemical, spills of lead compounds can expose researchers to levels of lead that can be harmful.(3) Lead(II) salts can be toxic and are suspected carcinogens.(4,5) Lead can also accumulate in the body through bioaccumulation in bone and other tissues.(6) It can cause a variety of well documented health problems and can even ultimately result in death.(7) Moreover, lead salts are most commonly provided in the form of fine powders that can easily become airborne and spread across large distances in the lab. Solvents which are commonly used to prepare perovskite formulations, such as dimethylformamide (DMF) and dimethyl sulfoxide (DMSO), can significantly exacerbate the intoxication of lead compounds as they are particularly effective in enhancing skin permeability, thus increasing the risk of absorption via dermal contact.(8) In addition, it is important to realize that contaminated clothing can also transport lead from the lab leading to secondary exposure. Therefore, controlling lead levels at the source is vital in order to reduce lead exposure.
The paper contains a nice table of "permissible" lead exposures:
 ...
...
...

The authors then make this less than reassuring statement:
The authors conduct tests using commercially available colorimetric test swabs and more quantitatively precise and accurate instrumentation, Inductively Coupled Plasma Mass Spectrometery (ICP-MS).
This table gives a flavor for the results:

They give a nice demonstration of how to change and remove one's gloves.
NOTE THAT THIS PROCEDURE IS PROBABLY QUITE USEFUL FOR CHANGING GLOVES POSSIBLY CONTAMINATED WITH COVID, AND FOR THAT MATTER, RADIOACTIVE MATERIALS.

The caption:
They conduct some cleaning experiments:

The caption:
The authors evaluate several commercial lead removal products, but note that the cleaning materials themselves represent a disposal problem.
They also give some scale to the production of 1 GW of lead perovskite solar cells:
By way of full disclosure, the institution where this work was performed was the King Abdullah University in Saudi Arabia. One of the authors, Ian McCulloch, holds a joint appointment at Oxford. I fully expect that some people will therefore assume that the work described herein is tainted. These are the same people who apparently believe that solar panels represent an alternative to the use of petroleum. This is a widely held belief, but it has never been effectively demonstrated anywhere on this planet. It is in fact, a myth, even though well over a trillion dollars has been expended in this century on the solar energy fantasy. In this century the use of oil has proceeded - in terms of energy produced - at three times the rate of wind, solar, geothermal and tidal energy combined, the growing by 34.79 exajoules to 188.45 exajoules, the latter, again combined by 9.76 exajoules to 12.27 exajoules. 12.27 exajoules is just about 2% of world energy demand.
2019 Edition of the World Energy Outlook Table 1.1 Page 38] (I have converted MTOE in the original table to the SI unit exajoules in this text.)
If I were Saudi and wanted to make the oil industry secure, I would encourage the further expenditure of money on solar cells, since they have proved ineffective entirely at displacing dangerous fossil fuels, as demonstrated by the fact, among many similar facts, that recently the arctic has burned owing to climate change, huge tracts of Australia have burned owing to climate change, and huge tracts of the American West Coast are currently burning owing to climate change. The rise in concentrations of the dangerous fossil fuel waste carbon dioxide is accelerating, and as of 2020, has reached an annual rate of 2.4 ppm/year.
Facts matter.
The authors recommend that laboratory work with perovskites be conducted in glove boxes and the precautions be taken to monitor the egress units. They recommend that international safety standards be established for laboratories working on lead perovskite materials.
There are, by the way, methods of safely dealing with hazardous waste from cleaning materials, paper towels, for example, and gloves, about which I've done considerable reading, but to my knowledge, they are not commercially practiced anywhere.
I trust you will have a pleasant Sunday as possible while respecting your own health and safety and those of others.
Sustainable Iron-Making Using Oxalic Acid: The Concept, A Brief Review of Key Reactions...
The paper I'll discuss in this post is this one: Sustainable Iron-Making Using Oxalic Acid: The Concept, A Brief Review of Key Reactions, and An Experimental Demonstration of the Iron-Making Process (Phatchada Santawaja, Shinji Kudo,* Aska Mori, Atsushi Tahara, Shusaku Asano, and Jun-ichiro Hayashi, ACS Sustainable Chem. Eng. 2020, 8, 35, 13292–13301)
There's a rumor going around that coal is dead, often accompanied with the delusional statement that so called "renewable energy" killed it. These statements are Trump scale lies. In the 21st century, has coal proved to be the fastest growing source source of energy, growing in terms of primary energy production by more than 63 exajoules from 2000 to 2018, faster than even dangerous natural gas, which was the next fastest growing source of primary energy, having grown by 50 exajoules from 2000 to 2018, roughly 700% percent faster and 600% faster than so called "renewable energy" in this period, ignoring biomass combustion, and hydroelectricity, the former being responsible for about 1/2 of the six to seven million air pollution deaths each year, the latter having destroyed pretty much every major riverine system in the world.
2019 Edition of the World Energy Outlook Table 1.1 Page 38] (I have converted MTOE in the original table to the SI unit exajoules in this text.)
One of the reasons that coal has grown so fast in this century is that poor people around the world - who basically we pretend don't exist - have not agreed to remain desperately impoverished so rich people in the post industrial Western world can pretend that their "Green" Tesla electric cars are powered by solar cells and wind turbines.
It would be a gross understatement to say that I am "skeptical" that so called "renewable energy" will do anything at all to address climate change. Half a century of cheering for it has done zero to prevent California, Australia, and indeed even the arctic from catching fire.
My least favorite form of so called "renewable energy" is represented by the wind industry, and one of my biggest criticisms of this benighted industry is its high mass intensity, particularly with respect to its high steel requirements, coupled with the short life time on average of wind turbines, typically well under twenty years.
There is essentially no steel that is not made on industrial scale with coal, today: Coal fires convert anthracite coal into coke, with the coke being used to reduce iron, alloying it with carbon, to make steel.
That's a fact. Facts matter.
Even if no energy were produced using dangerous fossil fuels - the use of which is rising, not falling - the problem of steel would remain, although it is possible that we may, to some extent, enter the age of titanium, are well into the age of aluminum, but the electrochemical reduction of both of these metals depend on carbon electrodes made from dangerous fossil fuels, coal coke and petroleum coke respectively.
This dependence is why this paper caught my eye.
From the introduction:
Extensive R&D efforts have been invested in alternative approaches to iron-making from the iron ore, which are largely classified into two types: direct reduction (DR) and smelting reduction processes.(2?6,9?12) In DR, the iron oxides in iron ore are reduced using reducing gas (H2 and CO) produced from natural gas or coal in reactors such as shaft furnaces and fluidized bed reactors. The reduction occurs at temperatures below the melting point of iron, producing so-called direct reduced iron or sponge iron. On the other hand, smelting reduction produces molten iron like a BF using a two-step process consisting of the solid-state reduction, followed by smelting reduction. The developed technologies, e.g., MIDREX for the DR and COREX, FINEX, ITmk3, and Hismelt for the smelting reduction, have been commercialized or are under demonstration.(3,6,13) The advantages of these alternative iron-making processes over BF include the lack of a need for coke, lower CO2 emissions, and lower capital/operation costs. However, they do not address the fundamental problems posed by the use of a BF because of their reliance on fossil fuels and harsh operating conditions. From this viewpoint, there are limited studies on potential sustainable iron-making methods...
There are many routes to oxalic acid with carbon dioxide as a starting material; one can come across papers along these lines regularly, some of which involve electrochemical reduction. Oxalic acid is moderately toxic, and is frequently utilized in commercial wood preservative products because it suppresses the viability of microorganisms that hydrolyze cellulose and lignin, the main constituents of wood. Famously the inability of the American Chestnut tree to synthesize oxalic acid when compared to the ability of the Chinese Chestnut to produce this biotoxin, led to the near extinction of the former. (Recently there has been promising work to insert oxalic acid generating genes into American Chestnuts.)
Oxalic acid is the simplest diacid, having the formula C2H2O4. It may be thought of as dimer formed by the elimination of two hydrogens from formic acid, the simplest carboxylic acid.
The overall scheme of this oxalic acid iron reduction scheme is shown in the following graphic from the paper:

The caption:
The authors note that among many acids designed to solvate iron oxides - which are clearly insoluble in water - is in fact oxalic acid, although mineral acids are more commonly utilized in this process.
They write:
...There are several factors affecting the rate of iron dissolution. Among them, pH of the initial solution, acid concentration, and temperature have been intensively studied.(17?24,26,28,30?32) The rate of iron dissolution is maximized when the pH of the oxalic acid solution is in the range of 2.5–3.0 because bioxalate anions (HC2O4–), which are responsible for iron dissolution, are the most abundant species in this range.(24,27,28) However, the pH of 2.5–3.0 is difficult to control with oxalic acid due to its low concentration, corresponding to 1–3 mmol/L, and, moreover, the low concentration is often insufficient for iron oxide removal. Therefore, the oxalic acid solution is typically prepared with the addition of its alkali salt as a buffering agent...
Dissolution is improved with the application of heat, which is unsurprising.
It is known that iron oxalate complexes can be reduced photochemically - this reaction has been used in actinometric devices - but the rate is slow, so the authors examine pyrolysis of the complex.
Figure 2:

The caption:
The authors avoid the hand waving "we're saved!" nonsense that often accompanies popular descriptions of lab scale processes when discussing the reduction of carbon dioxide to oxalic acid:
A robust approach to the reductive coupling of CO2 is electrochemical conversion.(56?58) For example, atmospheric CO2 is spontaneously captured and electrochemically converted into oxalate over copper complex, mimicking the natural photosynthetic transformation of CO2.(59) However, electrochemical conversion requires costly catalysts and organic solvents, which are unlikely candidates as an industrial method to produce cheap oxalic acid and iron. A recent report by Banerjee and Kanan(60) stood out in this regard, revealing the generation of oxalate only by heating cesium carbonate in the presence of pressurized H2 and CO2. The carbonate anion was replaced by formate anion from CO2. Then, the formate anion was coupled with CO2 to selectively form cesium oxalate with a yield of up to 56% (with respect to the carbonate), including other carboxylates at 320 °C and 60 bar. Nevertheless, the technical development of CO2 utilization for oxalic acid synthesis is still in its infancy.
In its infancy.
Given my personal focus on the utility of fission products, I note that hot cesium is one of the most prominent fission products, especially when freshly captured from used nuclear fuels. In addition, gamma radiation is known to produce carbon dioxide radicals, which may well accelerate this process.
But it's a very long way from here to there...
In any case, the authors experimentally (lab scale) use both photochemical and pyrolytic reduction of iron oxalate.
The following table shows the composition of the iron in each case.

XRD (X-ray diffraction) of the two processes:

The caption:
Scanning electron microscope (SEM) images of the product:

The caption:
In these graphics IO-A and IO-B refer to two different natural iron ores; CS refers too "converter slag" which represents iron recovered that would otherwise be waste.
Yields:

The caption:
A graphic representation of the dissolution process using oxalic acid:

All papers on processes have to make a stupid genuflection to the idea that solar energy will save the world, even though it won't:
The caption:
An attempt to build a plant around this idea would be to deliberately build a plant that is a stranded asset for large periods of a twenty four hour day, not to mention days when it rains, snows, or the sky is occluded by the smoke of uncontrolled fires because solar energy did not address climate change even after trillions of dollars and worldwide screams of cheering.

The caption:
On that score, a schematic of batch productivity:

The caption:
Excerpts of the conclusion and caveats against "We're saved!"
...A consideration is necessary for particle sizes of the feedstock iron ore, Fe(II) oxalate, and iron product. In the present experiment, fine particles of feedstock with sizes below 38 ?m were used to avoid possible influences of mass transfer on the iron dissolution. Fe(II) oxalates, obtained in the photochemical reduction, were also fine powders in the order of micrometers. In large-scale practical applications, technical difficulties would be found in feeding into and recovering from reactors for such small particles. Another concern is that small sizes of reduced iron product cause a low resistance to spontaneous ignition, which is also a problematic property of direct reduced iron.
The synthesis of oxalic acid from CO2 is vital to process sustainability. Direct synthesis is an emerging area of research but has a long way to go to become an industrial technology. Indirect synthesis via CO or biomass is a realistic option if a conversion system with economic and energetic rationality is found. It is also important to confirm the generation of CO2 and CO from iron-making, according to the proposed stoichiometry, and to design reactors that enable their recovery...
It's a cool paper on an area of research that I would certainly think is merited, not that anyone cares what I think.
Have a nice weekend. Please be safe and respect the safety of others.
Profile Information
Gender: MaleCurrent location: New Jersey
Member since: 2002
Number of posts: 33,512